一、簡介
嗜鉻細胞瘤(Pheochromocytoma)和副神經瘤(Paraganglioma)為一群會分泌Catecholamine的腫瘤,分別源發自腎上腺髓質以及交感神經節,兩者皆具有類似的臨床表現。
二、流行病學
→大部分的嗜鉻細胞瘤是偶發性的
→但有部分患者可能有家族遺傳性→患者年紀較小、且以雙側表現
要特別小心VHL、MEN2、NF1等疾病
三、臨床表現
(1) 典型的臨床表現
→Classic triad:陣發性頭痛、冒汗、心搏過速
→『5p』:Pressure (陣發性高血壓)、Pain(胸痛/頭痛)
Palpitation(心悸)、Perspiration(流汗)、Pallor(蒼白)
(2) 部分患者是沒有臨床表現,而是在影像檢查中意外發現(incidentoma)
(3) 腫瘤特性:10% tumor
10%雙側、10%家族遺傳、10%惡性、10%出現在小孩子
10%出現在腎上腺外
四、檢查與診斷
(1) 哪些人要想到Pheochromocytoma的可能性?
→出現典型症狀
→出現Hyperadrenergic spell (陣發性高血壓、冒汗、心悸、手抖等)
→年輕高血壓、難以控制的高血壓、相關家族史
→在腎上腺出意外發現的腫瘤(incidentoma)
(2) 相關檢查
a. 驗小便/血漿中的catecholamine以及其代謝產物
(代謝物包括Metanephrine和normetanephrine)
b. 2014年Endocrine Society guideline:
→一開始先驗24小時小便的fractionated metanephrines
→或是驗血漿中的metanephrine
c. 影像學:
→如果上述檢查懷疑有嗜鉻細胞瘤的可能,安排CT或MRI
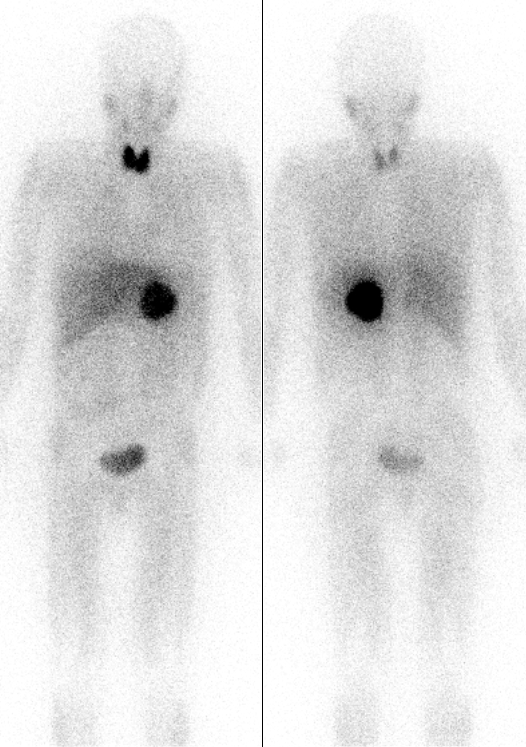
→核醫檢查:MIBG
(a) 如果CT/MRI沒看到,但是臨床上仍高度懷疑有嗜鉻細胞瘤
→此時可以安排MIBG核醫檢查
(b) MIBG為nonepinephrine類似物→會被adrenergic tissue攝入
(c) 在巨大(>10cm)的嗜鉻細胞瘤上,可以考慮安排
→因為惡性轉移的風險相對比較高
→FDG-PET:在轉移的病灶上,比CT/MRI/MIBG更為敏感
d. 基因檢測:當懷疑有家族遺傳性,考慮做基因檢測
五、治療
(1) 嗜鉻細胞瘤的治療,最重要是進行手術切除
(2) 術前準備:
→嗜鉻細胞瘤相當敏感,手術可能會誘發Catecholamine大量分泌
造成血壓飆升(導致hypertensive crisis),增加手術的風險
→術前準備的藥物治療目標
a. 控制高血壓、避免術中出現hypertensive crisis
b. 增加volume
→血壓控制:
a. 同時給alpha和beta blocker
→要先給alpha blocker,穩定後才給beta blocker
→alpha receptor會促進血管的收縮
若先給beta-blocker,阻斷後將會導致血管收縮→血壓會升高
b. 密切監控血壓
(3) 手術:adrenalectomy
(4) 惡性的pheochromocytoma:占10%
a. 局部:切除、放射治療
b. 系統性:
→放射性核種治療:I131 – MIBG:對於會吸收MIBG的腫瘤比較有效
→化學治療:Cyclophosphamide + Vincristine + Dacarbazine
Young WF, Kaplan NM (2016). Clinical presentation and diagnosis of pheochromocytoma. Retrieved 2016 Mar 18thfrom www.uptodate.com
Young WF, Kaplan NM, Kebebew E (2015). Treatment of pheochromocytoma in adults. Retrieved 2016 Mar 18th from www.uptodate.com
沒有留言:
張貼留言